In an invited presentation at the 2014 SIAM Conference on the Life Sciences, Norman Mazer began by rewording the question in the subtitle: Is a larger amount of cholesterol in high-density lipoprotein (HDL) particles invariably associated with a smaller risk of cardiovascular disease? He cited epidemiological studies showing that individuals with higher endogenous plasma levels of HDL cholesterol (HDL-C) have statistically lower risk of cardiovascular disease [2]. By contrast, he pointed to a number of recent clinical studies in which cholesteryl-ester transfer protein (CETP) inhibitors (and other compounds) were administered to raise HDL-C levels; those studies found the risk of cardiovascular disease to be either unchanged or increased in comparison to placebo treatments [1,6].
To understand the apparent paradox of HDL-C–raising therapy, Mazer and colleagues James Lu, Katrin Hübner, M. Nazeem Nanjee, and Eliot Brinton developed the LMK model of lipoprotein metabolism and kinetics [3]. Their model-based answer to the question is that raising HDL-C levels is “good” when it is associated with an increase in the rate of reverse cholesterol transport (RCT), the rate at which cholesterol is transferred from peripheral tissues to HDL particles in the bloodstream. The LMK model further showed that not all HDL-C–raising therapies raise the RCT rate.
The schematic of the LMK model (Figure 1) illustrates the steps in the RCT pathway. In the first step the ABCA1 transporter loads cellular cholesterol and phospholipid molecules onto lipid-poor ApoA-I protein dimers to create nascent discs. The discs are then converted to nascent spheres by the LCAT enzyme, which esterifies cholesterol molecules to create a lipid core within the particle. The nascent spheres typically fuse with the mature \(\alpha\)-HDL particles, which undergo a number of metabolic processes that either remove cholesteryl-ester (CE) molecules from the lipid core (SRB1 pathway), transfer CE to VLDL and LDL particles (CETP pathway), or remove entire HDL particles from the plasma (holo-particle uptake pathway). As a consequence of resulting changes in the surface-to-volume ratio of the HDL particles, excess ApoA-I in the surface is shed from the particles to regenerate lipid-poor ApoA-I (the HDL remodeling flux).
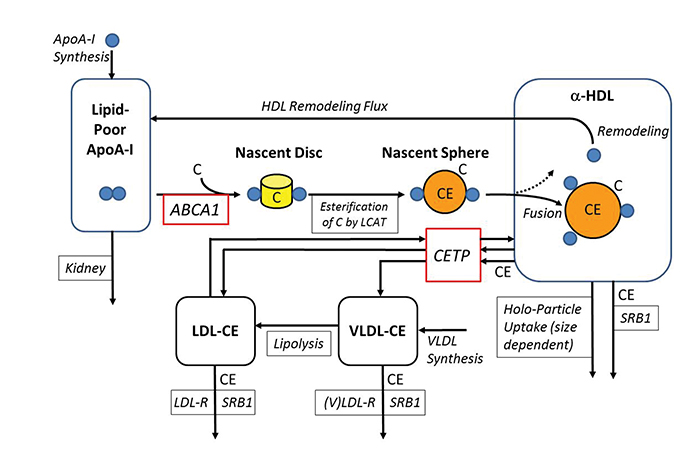
Figure 1. Schematic diagram of LMK model by J. Lu et al. [3]. See text for details of HDL formation and HDL-C–raising mechanisms (CETP inhibition and ABCA1 up-regulation).
|
The LMK model represents these various processes in eight ordinary differential equations with 16 parameters (Figure 2). Equations (1)–(3) describe the kinetic processes for ApoA-I in its lipid-poor and \(\alpha\)-HDL states; (4) and (5) describe the fusion of the nascent spheres with mature \(\alpha\)-HDL particles and the size-dependent holo-particle uptake mechanism; (6)–(8) describe the CE fluxes in the system. The key challenge in developing the LMK model, Mazer said, is representing the remodeling flux of ApoA-I by (3), which is based on the geometrical constraints imposed by the updated Shen model of HDL structure [3,5]. The group derived prior estimates of 14 of the model parameters from various literature sources. They obtained posterior estimates of all 16 parameters by calibrating the model to lipoprotein data from healthy subjects and from patients with heterozygous and homozygous CETP deficiency, using the maximum a posteriori method [3], thereby producing the parameter values for a nominal healthy subject.

Figure 2. Equations and parameters (numbered in red) in the LMK model; details can be found in [3].
|
To validate the model the group showed that it correctly predicted the effects of ABCA1 and ApoA-I gene mutations on HDL-C and ApoA-I levels and reproduced the bi-phasic kinetics observed in ApoA-I tracer kinetic experiments [3]. From the implementation perspective, Mazer noted that the ApoA-I tracer experiment can be simulated by adding an imaginary amount of ApoA-I \((i \times h)\) into the lipid-poor ApoA-I compartment, using MATLAB functions to numerically compute the analytic continuation of the model over the complex plane and dividing the imaginary part of the total ApoA-I level by \(h\). He credited Lu for the application of this novel approach, which is analogous to computing the derivative of the ApoA-I concentration by a complex-step derivative approximation [4].
Figure 3. Simulation of the relationship between the whole-body RCT rate and HDL-C in a virtual population (n=2000; blue). A subset of virtual subjects with HDL-C < 40 mg/dL (n=390; green) was subjected to 80% CETP inhibition (top) and 100% ABCA1 up-regulation (bottom; red). The purple dashed lines connect the asterisks with the X's for each individual subject during the respective treatments. While both treatments raise HDL-C levels by about 50%, CETP inhibition results in a slight decrease of RCT rate; ABCA1 up-regulation raises RCT rate markedly following the trend of the underlying virtual population.
The key insights provided by the LMK model on HDL-C–raising therapy were based on simulation of the relationship between the whole-body RCT rate and the HDL-C concentration in a virtual population of 2000 subjects (Figure 3). In this virtual population (whose model parameters were drawn from a multivariate normal distribution, with a standard deviation of 15% of the nominal subject’s values), a strong positive correlation was observed between the RCT rate and HDL-C (r = 0.95). Based on the assumption that the removal of cholesterol from atherosclerotic plaque lowers the risk of cardiovascular disease and that its rate parallels the whole-body RCT rate, the LMK model simulation would be consistent with the inverse relationship between HDL-C levels and cardiovascular disease risk seen in the epidemiological studies [2].
The group then used the model to simulate two different HDL-C–raising therapies in the subset of virtual subjects with HDL-C levels below 40 mg/dL. In the first case (Figure 3, top), inhibition of CETP activity by 80% was found to increase HDL-C levels by approximately 50%; the simulated RCT rates decreased slightly, however, rather than following the trend of the underlying virtual population. In the second case (Figure 3, bottom), up-regulating ABCA1 transporter activity by 100% also increased the HDL-C levels by about 50%; the RCT rates increased markedly, however, consistent with the trend of the underlying virtual population. Based on these simulations, the model suggests that CETP inhibition would neither increase RCT nor lower cardiovascular disease risk by this mechanism, whereas ABCA1 up-regulation would be expected to raise RCT rate and lower cardiovascular disease risk accordingly.
Mazer concluded that the LMK model provides a quantitative tool for evaluating the effects of different HDL-C–raising therapies and suggested that despite recent failures, some HDL-C–raising therapies may have the potential to lower cardiovascular disease risk.
Acknowledgments: The invited presentation and this article are dedicated to Ed Block, a founding member of SIAM and longtime friend of the speaker’s parents, Edy and Bob Mazer.
References
[1] P.J. Barter, M. Caulfield, M. Eriksson, et al., ILLUMINATE Investigators, Effects of torcetrapib in patients at high risk for coronary events, N. Engl. J. Med., 357:21 (2007), 2109–2122.
[2] Emerging Risk Factors Collaboration, Major lipids, apolipoproteins, and risk of vascular disease, JAMA, 302:18 (2009), 1993–2000.
[3] J. Lu, K. Hübner, M.N. Nanjee, E.A. Brinton, and N.A. Mazer, An in-silico model of lipoprotein metabolism and kinetics for the evaluation of targets and biomarkers in the reverse cholesterol transport pathway, PLoS Comput. Biol., 10:3 (2014), e1003509.
[4] J.R.R.A. Martins, P. Sturdza, and J.J. Alonso, The complex-step derivative approximation, ACM Trans. Math. Software, 29:3 (2003), 245–262.
[5] N.A. Mazer, F. Giulianini, N.P. Paynter, P. Jordan, and S. Mora, A comparison of the theoretical relationship between HDL size and the ratio of HDL cholesterol to apolipoprotein A-I with experimental results from the Women’s Health Study, Clin. Chem., 59:6 (2013), 949–958; doi: 10.1373/clinchem.2012.196949.
[6] G.G. Schwartz, A.G. Olsson, M. Abt, et al., dal-OUTCOMES Investigators, Effects of dalcetrapib in patients with a recent acute coronary syndrome, N. Engl. J. Med., 367:22 (2012), 2089–2099; doi: 10.1056/NEJMoa1206797.
Norman Mazer is the Disease Modeling Leader in Ophthalmology & Rare Diseases, Clinical Pharmacology, at Roche Pharma Research and Early Development, Roche Innovation Center, Basel, Switzerland. An interview conducted at the SIAM meeting can be seen here.